蛋白質摺疊問題一直是一個耗費巨大的難題,但是這個難題的解決又對人類具有巨大的意義。於是各個研究機構都開始尋找蛋白質摺疊問題的不同解,希望找到一種高效、準確的方式來解決這一難題。
幸運的是,在今年的 CASP(Critical Assessment of Techniques for Protein Structure Prediction)上,DeepMind 在今年找到了這個問題的其中一個「機器學習解」——AlphaFold,AlphaFold 的提出很好的解決了這一難題(在今年的 CASP 中獲得了第一名),同時又將 AI 所涉及的領域擴展到了生物科學。今天這篇文章就主要介紹機器學習是如何在生物科學中大放異彩的。本文首先介紹了蛋白質以及蛋白質領域目前面對的難題,然後介紹了機器學習能在這一領域做出什麼貢獻,最後對這一領域的問題及未來進行了展望。
什麼是蛋白質
蛋白質在我們的生命活動中扮演了極其重要的角色。幾乎我們身體的各項動作——收縮肌肉,感應光線或將食物轉化爲能量,都可以歸功於一種或多種蛋白質的配合。當你正在讀這篇文章的時候,你血液裏的血紅蛋白正在將氧氣運送到肌肉中,轉運蛋白正在爲神經元運送鈉以產生動作電位,而你之所以能讀懂這句話,眼睛裏的感光蛋白功不可沒。
不止這些「土生土長」的蛋白質在發揮作用,人工開發的蛋白質也已經被拼接到細菌基因組中以產生胰島素,或是分解塑料廢物來生產洗衣粉。所以,瞭解如何開發合適的人工蛋白質可以幫助我們提高生產效率,並開發具有全新功能的蛋白質。中學時期,我們其實已經對蛋白質有過不深的瞭解了。但是可能對很多人來說,高中已經很久遠了,爲了方便大家更好的理解本文,本節就對本文所需的蛋白質知識進行簡要介紹。
一般來說,總共有 20 種氨基酸,而蛋白質則是一條氨基酸鏈,它通過共價鍵將氨基酸連在一起。我們可以把氨基酸看作英文中的字母,而這個「字母」使我們可以將蛋白質表示爲一系列離散的標記,就像我們英語句子一樣。這種離散的順序表示形式被稱爲蛋白質的一級結構(Primary structure)。
然而,在細胞中,蛋白質是以三維結構存在的。由於蛋白質的功能與這個結構息息相關,因此瞭解這種 3D 結構極其重要。蛋白質的局部幾何結構稱爲二級結構(Secondary structure),這個結構也相應的決定了這一部分的特徵。最後,蛋白質的整體幾何結構稱爲三級結構(Tertiary structure),它決定了蛋白質的整體特徵。這些結構都是由 DNA 中的信息編碼的。
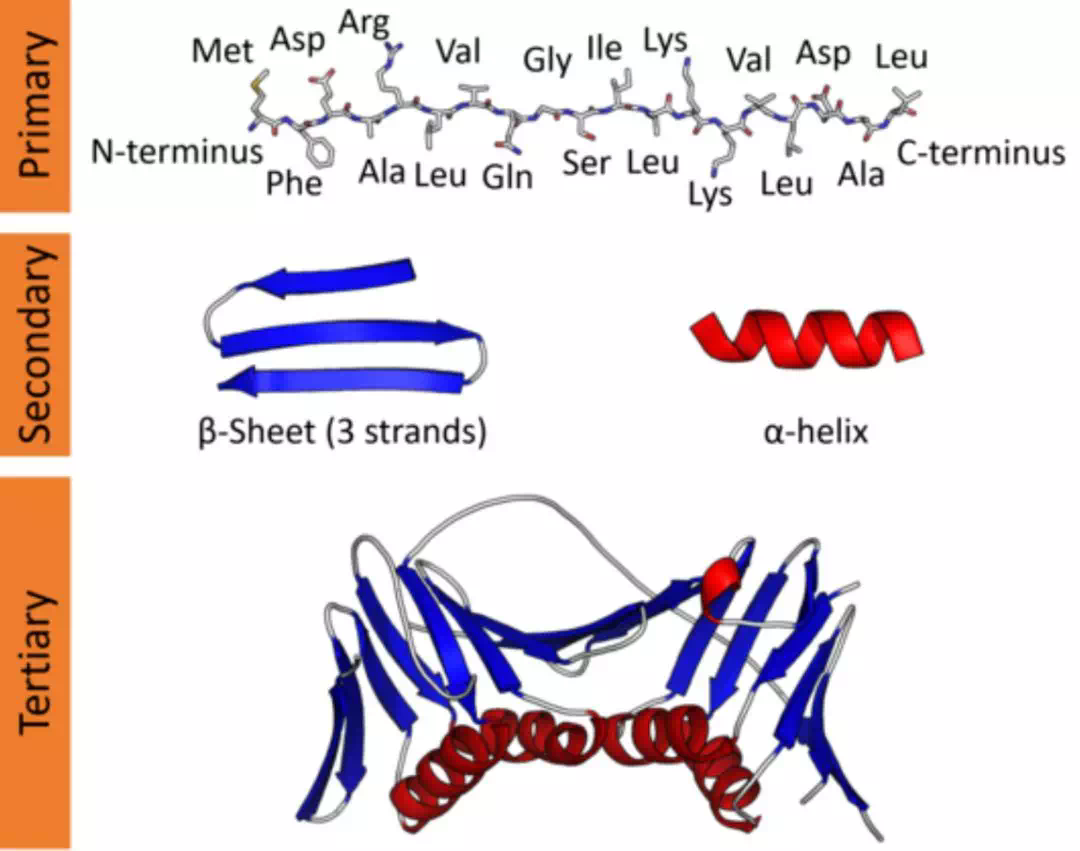
圖 1: 蛋白質的三種表達形式(圖源:https://bair.berkeley.edu/blog/2019/11/04/proteins/)
蛋白質摺疊問題
如上文所說,各種蛋白質的作用都取決於其獨特的 3D 結構。例如,構成我們免疫系統的抗體蛋白爲「Y 形」,類似於獨特的鉤子,通過鎖定病毒和細菌,這種蛋白能夠檢測並標記致病微生物以對這些病毒進行消滅。而膠原蛋白的形狀像繩索,可以在軟骨、韌帶、骨頭和皮膚之間傳遞張力。
其他類型的蛋白質也有很多,比如 Cas9,它以 CRISPR 序列爲指導,作用類似於見到,可以剪切和粘貼 DNA 片段;抗凍蛋白,其 3D 結構使它們能夠與冰晶結合並防止生物凍結;核糖體的功能則類似於程序化的裝配線,可幫助自身構建蛋白質。
因此,確定蛋白質的 3D 結構是非常重要的。如前文所述,3D 結構的確定,可以讓人們對蛋白質在體內的作用有更深入的瞭解,這樣科學家就能更有針對性地設計出有效的新療法。同時,對於一些由於蛋白質錯誤摺疊而引起的疾病(例如阿爾茨海默氏症,帕金森氏症,亨廷頓氏症和囊性纖維化等),瞭解了蛋白質正確的 3D 結構可以大大方便醫生對這些疾病的診斷和治療。
不僅如此,隨着越來越多的 3D 結構被確定,它也成爲藥品開發的隱藏力量。除了醫學外,蛋白質還可以有更大的貢獻,譬如幫助我們用對環境更友好的方式分解廢物的可生物降解酶就是通過蛋白質設計實現的,這種酶可以分解塑料和石油等污染物。雖然 3D 結構的確定可以給我們帶來這麼多好處,但是蛋白質越大,建模越複雜和困難,因爲要考慮的氨基酸之間存在更多的相互作用。正如列文塔爾悖論中指出的那樣,枚舉典型蛋白質的所有可能構型所花費的時間要比宇宙的年齡長,才能達到正確的 3D 結構。
而且,僅從蛋白質的基因序列(一級結構)中找出蛋白質的 3D 形狀(二、三級結構)是一項極其複雜的任務。不幸的是,經過數十年的研究,科學家們也都發現了這個難題是無法被繞開的——DNA 僅包含蛋白質的一級結構信息,卻並不能探測到這些蛋白質是如何摺疊的(3D 結構是如何的)。
這時問題也就提出來了,即所謂的「蛋白質摺疊問題」——預測這些鏈(一級結構)是如何摺疊成複雜的 3D 結構的。爲了促進研究和衡量最新方法以提高預測的準確性,1994 年成立了兩年一度的全球競賽,名爲「Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction」(CASP),這一競賽現已成爲用於評估技術的通用標準。
爲什麼要引入機器學習?
在過去的五十年中,科學家們已經能夠使用諸如冷凍電子顯微鏡(cryo-electron microscopy)、核磁共振(nuclear magnetic resonance)、X 射線晶體學(X-ray crystallography)之類的實驗技術確定蛋白質的形狀,但是每種方法都需要大量的時間和精力來做實驗。爲了發現一個結構,可能需要數年的時間以及數萬美元的投資。這就是爲什麼生物學家開始將目光轉向 AI,希望 AI 能找到合適的方法來替代這一漫長而費力的工作。
幸運的是,由於基因測序成本的快速降低,基因組學領域的數據非常豐富,對應的序列的數量也在呈指數增長。
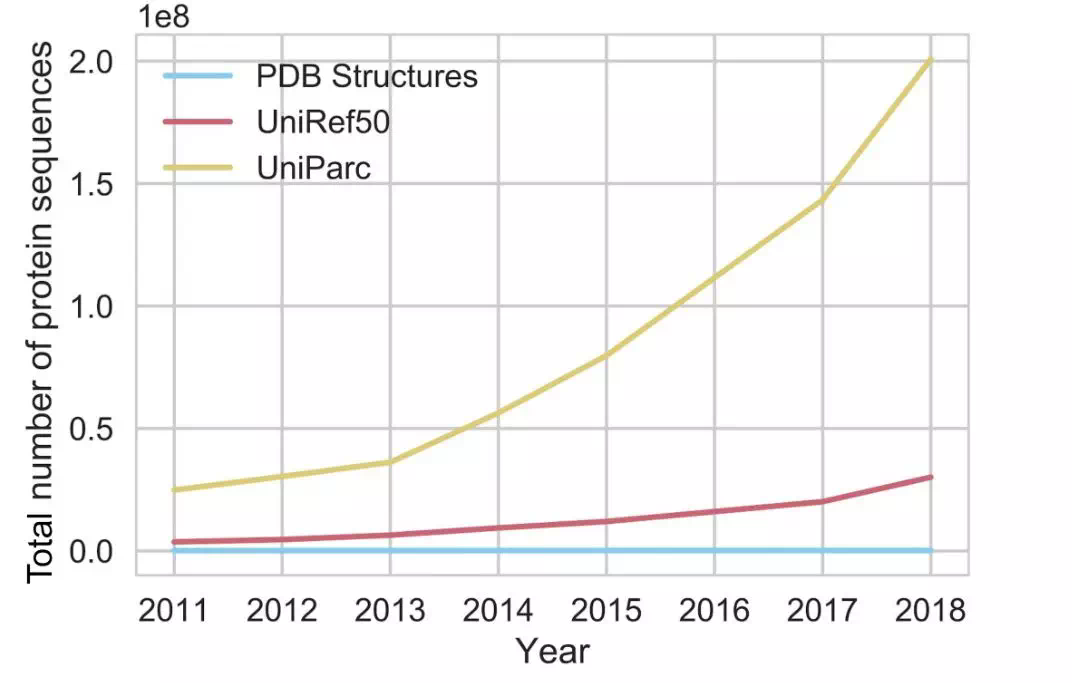
圖 2:蛋白質序列數目變化(圖源:https://bair.berkeley.edu/blog/2019/11/04/proteins/)
因此,在最近幾年中,依賴於基因組數據的用於預測問題的深度學習方法變得越來越流行。DeepMind 也開始介入這個困難的問題,並基於此研發出了 AlphaFold,並被 CASP 組織者誇讚爲「前所未有的進步」。
現有數據中的寶藏
既然要引入機器學習,首先要確定的問題就是該用什麼機器學習技術呢。如果要對這個問題進行監督學習,就需要標籤。在這個問題中,我們就需要標記蛋白質中每個原子的三維座標。給蛋白質貼標籤是一個勞動密集型、資源密集型且耗時巨大的過程,而且這個過程只能由專家來完成,具體來說,只能由使用價值 1 萬美元/小時的顯微鏡的專家來完成。因此監督學習這條路目前來說是走不通的。
如前文所說,雖然有標記的數據很少,但是沒有標記的基因組數據(蛋白質序列)是很多的,如果要更快更好的完成任務,顯然第一個要做的就是學會使用這些數據。事實證明,這些數據中包含的有用信息是很多的,進化關係(evolutionary relationship)就是其中一個。進化關係的本質就是同源性,同一個祖先(家族)的兄弟應該具有相似的特性。爲了更好的理解什麼是進化關係,下面先對當下科學家們提取這一關係的主要方式——序列對齊法——進行簡要介紹。
在這個方法中,我們將要查詢序列放到整個數據庫中,尋找其「兄弟」序列(即同祖先的後代)。圖 3 就是一個簡單的匹配例子(這裏的 A, T(或 U), C, G 是指核酸,蛋白質匹配的情況更加複雜,後面會講)。這個例子中,我們可以用點(.)來代表少量的不匹配,用破折號(-)表示缺失值。

圖 3:序列匹配實例(圖源:https://en.wikipedia.org/wiki/Sequence_alignment)
在對序列匹配有了大概的瞭解後,再來看一下蛋白質的匹配是怎麼做的。除了要將核酸變成氨基酸外,還要注意其生物特性。下圖就是 GFP 蛋白家族的部分序列比對,這些蛋白質都是熒光蛋白,也就是可以發光。圖中的顏色代表不同的氨基酸組,同一個組內的氨基酸有相同的生物物理特性,也就被標成了相同的顏色。
具體來說,紫色代表疏水性(C,A,V,L,I,M,F,W),紅色代表帶電(D,E,R,K),綠色代表正極不帶電(S,T,N,Q)。如果兩個序列的顏色一致,也就表示這些位置的特性在進化過程中被保留了。
 圖 4:蛋白質序列匹配實例(圖源:https://bair.berkeley.edu/blog/2019/11/04/proteins/)
圖 4:蛋白質序列匹配實例(圖源:https://bair.berkeley.edu/blog/2019/11/04/proteins/)
所以我們到底想從進化關係中得到什麼呢?就如上圖的例子所示,這些蛋白質具有類似的功能(發光),但是有些位置是被保留的,有些位置卻不一樣了——進化是不確定的,並不是完全的繼承(顏色一模一樣)。有的時候這些顏色(氨基酸)的改變並不會引起蛋白質結構的改變,相應的,這個蛋白質的功能也不會改變。
因此,我們希望從數據庫中提取這樣的信息——進化在什麼位置是自由的,在什麼地方是有迴旋餘地的,在什麼地方是完全受束縛的。而這些信息,正是在做結構預測時的重要輸入。舉個簡單的例子,三維空間中臨近的位置一般會共同進化,即一個位置的突變通常也會引起相鄰位置的突變,如果突變沒有共同發生,那這兩個氨基酸應該不在相鄰的位置。
NLP 與蛋白質摺疊問題
大語料庫、難以獲得的標籤、序列對齊、嵌入、token 序列,再加上前文說過的要從中提取出的信息(嵌入),學習過自然語言處理的同學可能會感覺自己回到了自然語言處理(NLP)的課堂上,對那些 NLP 的研究者們來說,這種聯繫更是昭然若是。因此,研究者們開始將目光轉向 NLP 最近的重大突破之一——自監督學習,即從未標記數據中獲取有用信息的方法。這之中較爲傑出的代表就是 BERT 了,這裏先對 BERT 做簡單的介紹。
如下圖所示,在 BERT 訓練時,先遮住部分單詞,然後通過其他單詞來預測這個被擋住的單詞。這樣我們就可以得到一個關於可能成爲的單詞的分佈,然後通過交叉熵損失來訓練這個模型。學習後的模型可以從序列中學習到該序列的特性,並且這些特性(嵌入)可以很容易的被遷移到下游任務中去。
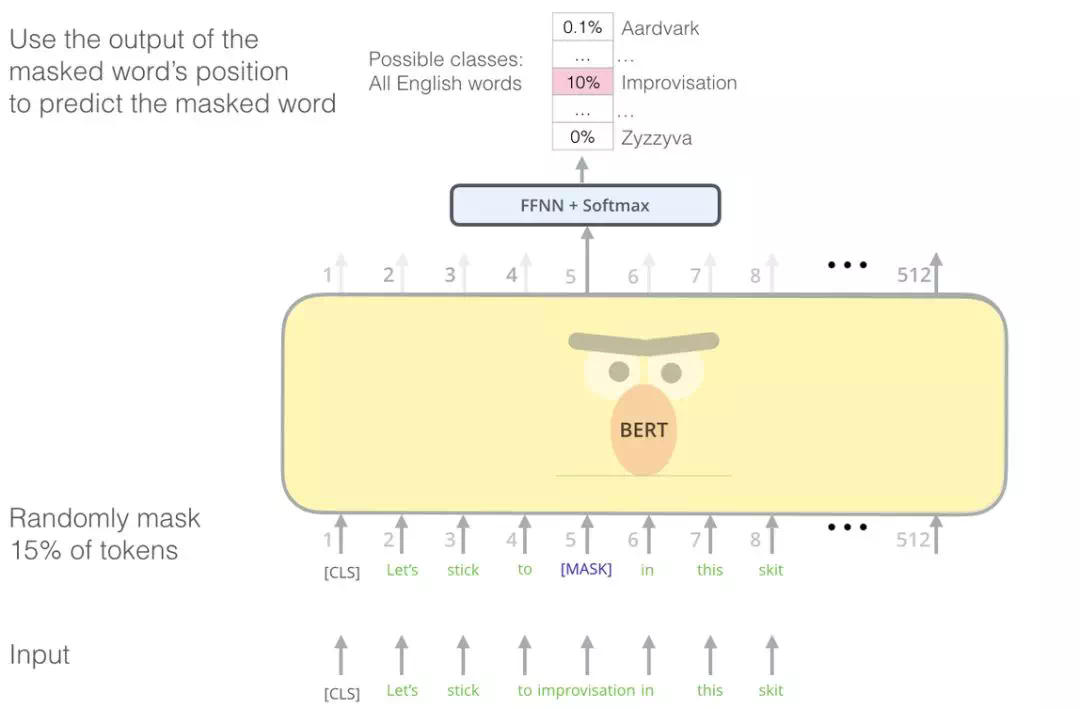
圖 5:BERT 流程圖(圖源:https://jalammar.github.io/illustrated-bert/)
那如果我們把句子變成氨基酸序列,下游任務變成蛋白質結構預測,是不是就變成了蛋白質摺疊問題的模型了?
這樣任務就變成了:輸入氨基酸序列,通過 BERT 獲得蛋白質嵌入,通過蛋白質嵌入預測蛋白質結構。
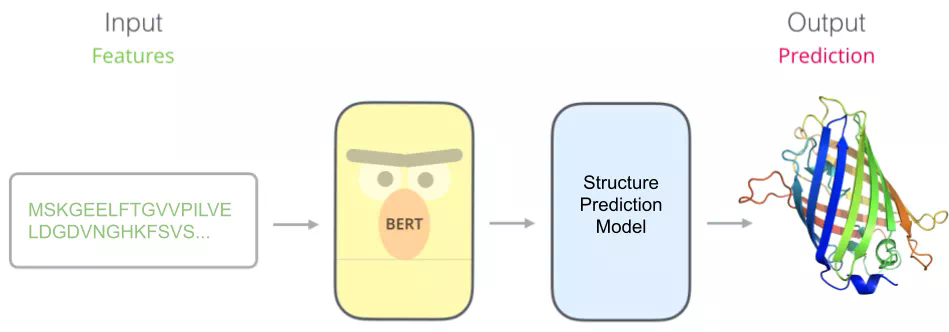
圖 6:蛋白質模型流程圖(圖源:https://bair.berkeley.edu/blog/2019/11/04/proteins/)
TAPE(Tasks Assessing Protein Embeddings)
理論已經說了很多了,那麼具體應該怎麼實施呢,這樣做到底效果如何呢?在《Evaluating Protein Transfer Learning with TAPE》中,就像 NLP 任務中有 GLUE 作爲 Benchmark 一樣,作者基於 NLP 任務中的 GLUE 提出了蛋白質嵌入的 benchmark——TAPE(Tasks Assessing Protein Embeddings),並使用多個深度學習模型在包括結構預測在內的多類下游任務(同源檢測,蛋白質工程)上進行了測試。
首先簡要介紹一下三個下游任務的意義。這三個下游任務中,結構預測已經在前面提到過了;因爲改變其氨基酸組成 (即改變序列的一個字母) 會改變蛋白質的性質,大多修改都會破壞其性質,而且跟原序列相差越大,就越不可能維持一個蛋白質的功能,因此蛋白質工程問題解決的則是如何對蛋白質進行何種修改以優化特定的功能的問題(比如讓一種熒光蛋白髮出的光更亮);而同源檢測任務則是發現兩個蛋白質是否是同源的(前面提到的用序列對齊解決的問題)。這些任務都有各自的作用,比如蛋白質工程可以用於優化流感抗體的效力以製造更好的疫苗,或增加用於材料合成的生化催化劑的產量。
在 TAPE 中,作者使用了兩個 NLP 自監督任務中常見的損失函數。第一個 next-token 預測任務中的損失函數,它評測了 p(x_i |x_1,…,x_i - 1)。但是很多蛋白質任務是 seq2seq 的,需要雙向的上下文信息,因此作者加入了反向模型,即 p(x_i |x_i+1,…,x_L),這個每個位置都有雙向上下文信息了。第二個是 Masked-token 預測任務中的損失函數,它評測了 p(x| x_unmask)。同時,作者還使用了一種專門應用於蛋白質任務的損失函數,即有監督預訓練任務損失。
在模型的選擇上,作者使用了 Transformer,LSTM 和 Dilated ResNet。Transformer 有 12 層,每一層有 512 個隱藏單元和 8 個注意頭,這樣這個模型就有 3800 萬個參數。LSTM 則由兩個三層 LSTM 組成,與 ELMO 類似,對應於正向和反向語言模型,它有 1024 個隱藏單元,這些輸出會在最後一層被連在一起。ResNet 中由 35 個 Res 塊,每個塊包含兩個卷積層和 256 個過濾器,內核大小爲 9,擴展率(dilation rate)爲 2。三個模型的超參數量都基本相同。
TAPE 使用 Pfam(3100 萬數據)作爲預訓練的訓練集,這裏面的數據根據進化關係被聚類成不同的家族。同時,在下游任務中,作者使用了 5 個數據集(對應前面說的 3 類下游任務),數據集大小如下表所示。
表 1:數據集大小(圖源:https://www.biorxiv.org/content/10.1101/676825v1.full)
在分訓練集和測試集時使用兩種方式——隨機分組(Random Families)和根據家族進行分組(家族分組,Heldout Families)。作者使用準確性(Accuracy)、複雜性(Perplexity)和指數交叉熵 (ECE) 作爲評價指標,在語言模型訓練任務中,對使用自學習訓練的 3 個模型(前三個)進行了評測,並與過去提出的有監督模型以及一個隨機的 baseline 模型進行了對比,結果如下表所示。
表 2:預訓練評測結果(圖源:https://www.biorxiv.org/content/10.1101/676825v1.full)
從表中可以看出,通過自學習訓練出的模型的精度(越高越好)和 ECE(越低越好)都優於其他模型,而家族分組精度始終低於隨機分割精度,顯示出其泛化能力的下降。要注意的是,儘管一些模型在隨機分割集和輔助集上的 perplexity 都比其他模型低,但這種較低的 perplexity 並不一定對應於下游任務的更好性能。
同時,作者也對這些模型在 5 項任務上的表現進行了評測。這 5 項任務分別是:
二級結構預測(Secondary Structure (SS) Prediction -Structure Prediction Task ):屬於結構預測任務,顧名思義,預測蛋白質的二級結構,是一個 seq2seq 任務,每一個氨基酸會獲得一個 label(螺旋或是鏈裝)。評價標準是準確率。
圖 7:結構預測、關聯預測、遠程同源預測(圖源:https://www.biorxiv.org/content/10.1101/676825v1.full)
關聯預測(Contact Prediction - Structure Prediction Task):屬於結構預測任務,對輸入的氨基酸進行配對(根據一定原則),如果兩個氨基酸被判斷爲關聯(in contact),則其關係標記爲 1,反之標記爲 0。評價時採用結果中前 5 的精確率。
遠程同源檢測(Remote Homology Detection - Evolutionary Understanding Task):屬於同源檢測任務,將輸入的蛋白質序列映射到特定的摺疊結構上去。本質上是序列分類任務,最終採取準確率作爲測量標準。
熒光度預測(Fluorescence Landscape Prediction - Protein Engineering Task):屬於蛋白質工程,本質上是迴歸任務。如圖 (a) 所示,將每個蛋白質映射到一個熒光度上。測試時,使用 Spearman 提出的ρ係數(評價相關度)作爲指標。
穩定度預測(Stability Landscape Prediction - Protein Engineering Task):也屬於蛋白質工程,跟熒光度預測類似,預測內容爲某個氨基酸爲了保持穩定所需要保持的最大範圍。
圖 8:熒光度預測、穩定度預測(圖源:https://www.biorxiv.org/content/10.1101/676825v1.full)
下表記錄了各項任務中各個方案的表現,表中的數據根據任務不同,有可能是準確率(accuracy)、精確度(precision)或是 Spearman』s ρ,但是都是越大越好。可以看到預訓練對大部分任務的提升是很明顯的。

表 3:預訓練與其他方法對比(圖源:https://www.biorxiv.org/content/10.1101/676825v1.full)
AlphaFold
最後再回到將我們視線聚焦到蛋白質領域的 AlphaFold,DeepMind 具體做了什麼呢?AlphaFold 其實是深度學習方法與傳統方法的結合,具體來說就是 CNN+Rosette,本文主要介紹深度學習相關的部分(CNN),對傳統方法(Rosette,也是一個很優秀的算法,在 AlphaFold 奪冠的過程中功不可沒)就不做詳細介紹了,有興趣可以自己瞭解一下。
在 AlphaFold 中,他們一共設計了三個不同的方法:首先是最傳統的方法,跟從前的專家系統極其相似——先從一維的氨基酸序列生成一個二維的接觸距離矩陣(contact matrix)以記錄兩個氨基酸之間的距離。然後把蛋白質分割成幾個結構域(domain)——一個 domain 內的的相互接觸很強,但是 domain 內的分子和其之外的接觸則相對較弱。然後預測蛋白質骨架的摺疊角度,根據蛋白質骨架的摺疊角度,把結構切割成一系列有重疊的 9 個氨基酸殘基爲單位的短肽,分別預測,再組裝到一起,預測整個結構域的結構。最後,把摺疊好的結構組裝到一起。
這個方法就是簡單的從頭預測(ab initio)方法的框架,在每一步都有一個評分系統,從而保留好結果,刪除差結果——沒有深度學習的時候也是這麼做的。方法 2 則在方法 1 的基礎上,不再「切割成小的短鏈分別預測」,而是直接預測整個結構域。方法 3 在方法 2 的基礎上,又去掉了結構域分割這一步,這個時候步驟就變成了——從一維的氨基酸序列生成一個 2 維的接觸距離矩陣,預測蛋白質骨架的摺疊角度,最後梯度下降出結果。
這裏用到深度學習的地方主要有三個:第一個用到深度學習的地方是 contact matrix 預測。這裏使用了一個卷積網絡 CNN 把一維的氨基酸序列,展成 contact matrix。訓練這個 contact matrix 網絡時首先進行了 BLAST 序列比對,然後通過序列比對得到的特徵進行預測。這個網絡深度應該非常高,但其實今年早些時候的 DNCON2 這種淺層的也可以做。網絡中會用 likelihood 作爲估分方式,先進行一次篩選。
第二個用到深度學習的地方是在預測蛋白質骨架結構的時候,這裏要描繪每個鍵平面之問的二面角 torsion angle,這一步直接調了以前的一個圖像生成的方法——A Recurrent Neural Network For Image Generation。第三個就是每一步的評估網絡,這個評估網絡也是一個 CNN。輸入評估網絡的是第一個 CNN 生成的 contact malrix,序列比對產生的特徵,還有結構的幾何結構之類的。
展望及挑戰
如前文所示,深度學習本身並不能很好的解決蛋白質預測問題,而且預訓練也沒有讓所有任務上的表現都變得更好,可以看到,在那些非監督學習的下游任務(如 Contact prediction 任務)中預訓練就表現得差了些。而在 CASP 中奪冠的 AlphaFold 雖然在一定程度上緩解了人工的壓力,但是卻對硬件有極高的要求,所以在一定程度上 AlphaFold 的勝利也可以說是 DeepMind 硬件的勝利,並沒有從根本上找到「機器學習」解。
但是 CASP 中,前 5 名都使用了深度學習技術,其他一些使用了深度學習技術的隊伍也取得了不錯的成績。因此,綜合 CASP 以及 TAPE 的結果來看,機器學習和自學習都是蛋白質預測未來的大方向。而且在自學習快速發展的當口,蛋白質也爲其提供了一個巨大的試驗場(擁有巨大的序列庫),因此我相信蛋白質任務和自學習會在相互促進的過程中發展越來越好。
作者介紹:本文作者爲王子嘉,目前在帝國理工學院人工智能碩士在讀。
參考文獻
AlphaFold: Using AI for scientific discovery. Accessed at: https://deepmind.com/blog/article/alphafold
Can We Learn the Language of Proteins? Accessed at: https://bair.berkeley.edu/blog/2019/11/04/proteins/
Illustrated Bert. Accessed at: https://jalammar.github.io/illustrated-bert/
如何看待 AlphaFold 在蛋白質預測領域的成功?Accessed at: https://www.zhihu.com/question/304484648