就在幾個月前,DeepMind推出了AlphaFold系統,這個被稱爲生物界「AlphaGo」的系統能夠預測並生成蛋白質3D結構。而近日,來自MIT的研究人員開發了一個新的研究模型,能夠直接預測氨基酸鏈片段是如何決定蛋白質功能的。這一發現可以幫助研究人員設計和測試新的蛋白質,從而用於藥物研發和生物學研究。
我們都知道,蛋白質是維持我們生命所必需的龐大而複雜的物質。蛋白質具體能完成什麼樣的功能,主要取決於它獨特的三維結構。因此瞭解蛋白質的結構,對於預測其對某些藥物的反應來說,是一個非常重要的環節。
▲每一個蛋白質都擁有錯綜複雜的三維結構(圖片來源:123RF)
然而,儘管有了數十年的研究和多種成像技術的輔助,我們仍然只瞭解到了無數蛋白質結構中的很小一部分,還有很多未知結構的蛋白質功能尚未揭曉。針對這一情況,來自MIT的研究人員開發出了一種方法,「學習」了蛋白質序列中每個氨基酸位置上容易計算的表徵。隨後,研究人員將這些表徵輸入機器學習模型,讓模型直接預測單個氨基酸片段的功能,而無需任何蛋白質結構的數據。
首先,研究人員使用了來自蛋白質結構分類數據庫(SCOP)的約22000種蛋白質,將這些蛋白質按照結構和氨基酸序列的相似性進行分類,並對機器學習模型進行訓練。對於每一對蛋白質,研究人員都會根據其SCOP類別計算出一個結構相似性評分。然後,研究人員將隨機的蛋白質結構對及其氨基酸序列輸入機器學習模型,通過編碼器將它們轉換成數值表示出來,稱爲嵌入(embedding)。每個嵌入都包含了一對氨基酸序列的相似性信息。
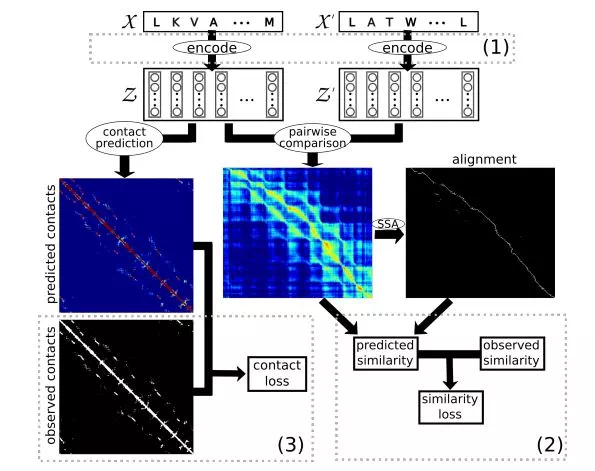
▲該模型的示意圖(圖片來源:arxiv.org)
該模型將兩個嵌入對齊,然後計算出相似度評分,以預測其代表的蛋白質三維結構的相似性。然後,計算機會將這一評分與真實的SCOP相似性評分進行比較,並向編碼器發送反饋信號。如果模型的預測分數與真實分數相差較遠,則會進行一定的調整。
同時,該模型預測了每次嵌入的「接觸圖」(contact map),即每個氨基酸與該蛋白質中其他氨基酸的距離,並將其預測的接觸圖與來自SCOP的已知接觸圖進行比較,然後向編碼器發送反饋信號。這一步驟有助於模型更好地明確氨基酸在蛋白質結構中的確切位置,從而進一步瞭解每個氨基酸的功能。
對於某個氨基酸鏈,該模型可以爲三維結構中的每個氨基酸位置生成一個嵌入。然後,機器學習模型可以使用這些序列嵌入,根據其預測的三維結構接觸圖,來準確預測每個氨基酸的功能。在一個應用實例中,研究人員使用該模型預測有哪些蛋白質可以通過細胞膜,其預測結果比現有的先進模型還要更加準確。
接下來,研究人員計劃將該模型應用到更多的預測任務中,例如弄清楚哪些序列片段可以與小分子結合,這對於藥物研發工作來說是至關重要的。研究人員表示,這項研究最終將可以應用於人類健康和藥物基因組學,因爲它有助於檢測破壞蛋白質結構的有害突變。
參考資料:
[1] Bepler, et al., (2019). Learning protein sequence embeddings using information from structure. ICLR 2019, arXiv:1902.08661
[2] MIT CSAIL’s AI predicts a protein’s function from chains of amino acids. Retrieved March 26, 2019, from https://venturebeat.com/2019/03/22/mit-csails-ai-predicts-a-proteins-function-from-chains-of-amino-acids/
[3] Model learns how individual amino acids determine protein function. Retrieved March 26, 2019, from http://news.mit.edu/2019/machine-learning-amino-acids-protein-function-0322